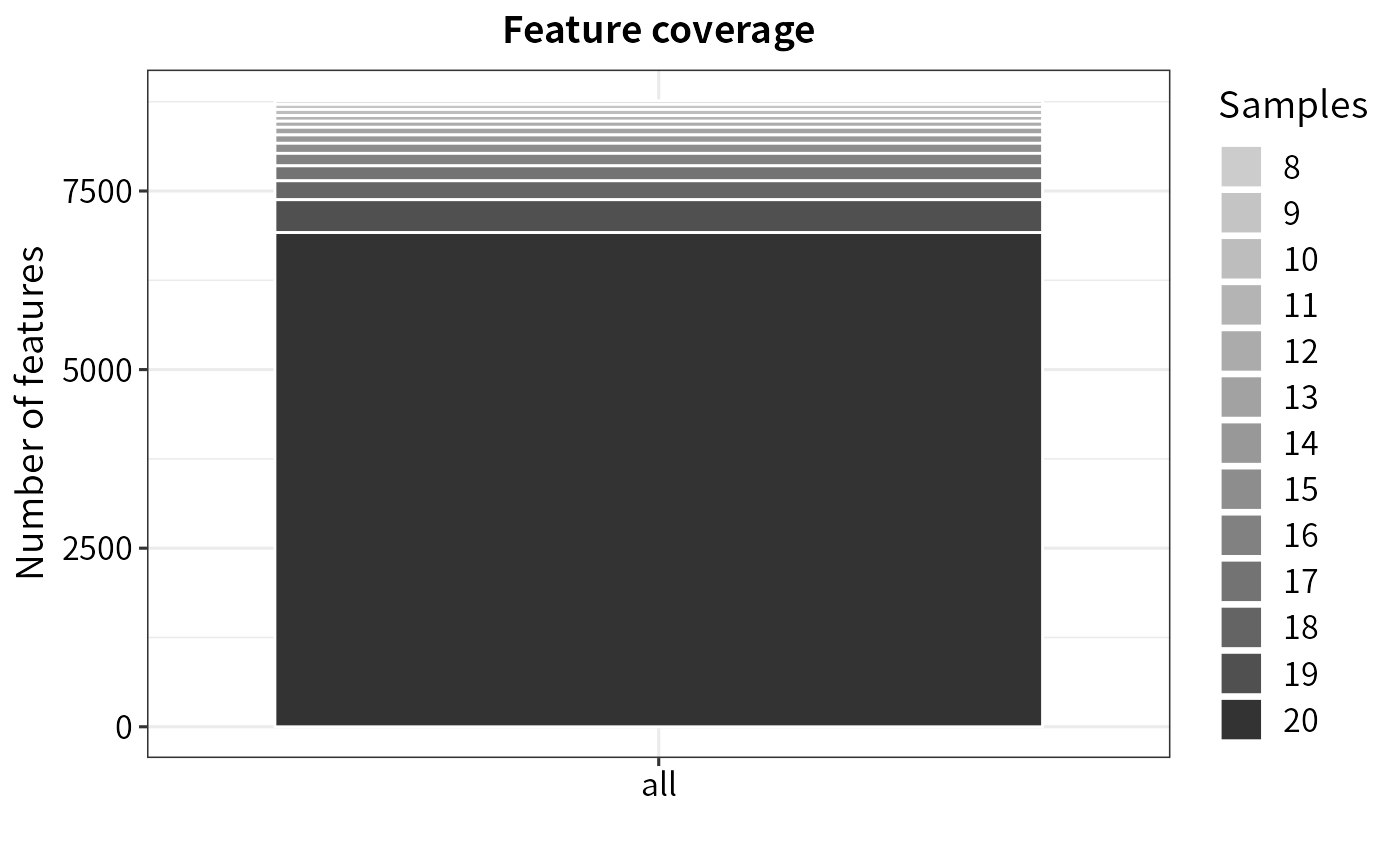
Plot protein coverage
plot_coverage.Rdplot_coverage generates a barplot
of the protein coverage in all samples.
Arguments
- se
SummarizedExperiment, Data object for which to plot observation frequency.
- plot
Logical(1), If
TRUE(default) the barplot is produced. Otherwise (ifFALSE), the data which the barplot is based on are returned.- features_type
Character(1), the type of features used in title, like 'proteins', 'genes', 'peptides', default is 'features'
Value
Barplot of protein coverage in samples
(generated by ggplot)
Examples
# Load example
data(Silicosis_pg)
data <- Silicosis_pg
data_unique <- make_unique(data, "Gene.names", "Protein.IDs", delim = ";")
# Make SummarizedExperiment
ecols <- grep("LFQ.", colnames(data_unique))
## Load experiement design
data(Silicosis_ExpDesign)
exp_design <- Silicosis_ExpDesign
se <- make_se(data_unique, ecols, exp_design)
# Filter and normalize
filt <- filter_se(se, thr = 0, fraction = 0.4, filter_formula = ~ Reverse != "+" & Potential.contaminant!="+")
#> filter base on missing number is <= 0 in at least one condition.
#> filter base on missing number fraction < 0.4 in each row
#> filter base on giving formula
plot_coverage(filt)