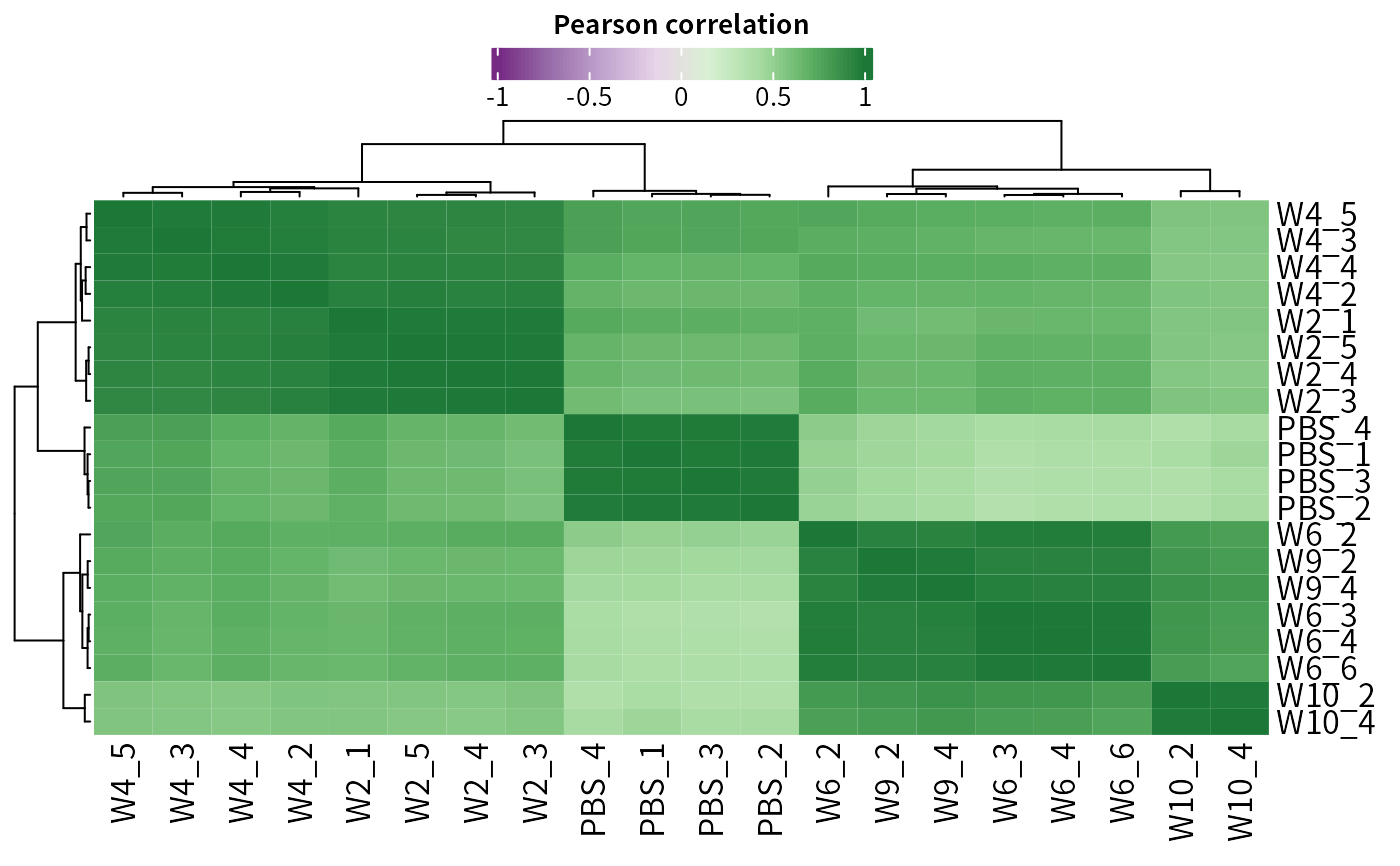
Plot correlation matrix
plot_cor.Rdplot_cor generates a Pearson correlation matrix.
Usage
plot_cor(
x,
significant = TRUE,
lower = -1,
upper = 1,
pal = "PRGn",
pal_rev = FALSE,
indicate = NULL,
font_size = 12,
plot = TRUE,
add_values = FALSE,
value_size = 10,
digits = 2,
...
)Arguments
- x
SummarizedExperiment, Data object for which differentially enriched proteins are annotated (output from
test_diff()andadd_rejections()).- significant
Logical(1), Whether or not to filter for significant proteins.
- lower
Integer(1), Sets the lower limit of the color scale.
- upper
Integer(1), Sets the upper limit of the color scale.
- pal
Character(1), Sets the color panel (from RColorBrewer).
- pal_rev
Logical(1), Whether or not to invert the color palette.
- indicate
Character, Sets additional annotation on the top of the heatmap based on columns from the experimental design (colData).
- font_size
Integer(1), Sets the size of the labels.
- plot
Logical(1), If
TRUE(default) the correlation matrix plot is produced. Otherwise (ifFALSE), the data which the correlation matrix plot is based on are returned.- add_values
Logical(1). Display the distance value on heatmap.
- value_size
Numeric(1). Font size of distance.
- digits
Integer(1), indicating the number of decimal places of distance.
- ...
Additional arguments for Heatmap function as depicted in
Heatmap
Value
A heatmap plot (generated by Heatmap)
Examples
# Load example
data(Silicosis_pg)
data <- Silicosis_pg
data_unique <- make_unique(data, "Gene.names", "Protein.IDs", delim = ";")
# Make SummarizedExperiment
ecols <- grep("LFQ.", colnames(data_unique))
## Load experiement design
data(Silicosis_ExpDesign)
exp_design <- Silicosis_ExpDesign
se <- make_se(data_unique, ecols, exp_design)
# Filter and normalize
filt <- filter_se(se, thr = 0, fraction = 0.4, filter_formula = ~ Reverse != "+" & Potential.contaminant!="+")
#> filter base on missing number is <= 0 in at least one condition.
#> filter base on missing number fraction < 0.4 in each row
#> filter base on giving formula
norm <- normalize_vsn(filt)
#> vsn2: 8762 x 20 matrix (1 stratum).
#> Please use 'meanSdPlot' to verify the fit.
# Impute missing values using different functions
imputed <- impute(norm, fun = "MinProb", q = 0.05)
#> Imputing along margin 2 (samples/columns).
#> [1] 0.3026531
# Test for differentially expressed proteins
diff <- test_diff(imputed, "control", "PBS")
#> Tested contrasts: W10_vs_PBS, W2_vs_PBS, W4_vs_PBS, W6_vs_PBS, W9_vs_PBS
#> Strimmer's qvalue(t)
dep <- add_rejections(diff, alpha = 0.05, lfc = 1)
# Plot correlation matrix
plot_cor(dep)